
研究背景
金属的溶解及其逆过程对腐蚀和电沉积都是不可或缺的一部分;然而,许多关于溶解过程的机理细节都难以破译。这些因素包括在电极电位下,金属和溶剂相互作用的竞争如何对离子溶解动力学和电荷转移的影响。劳伦斯利物莫国家实验室Shubham Sharma,Tuan Anh Pham和Brandon C. Wood 等人引入了一个基于密度泛函理论的计算框架和电子的大标准处理,直接预测了在恒定势下金属溶解的势能景观。以铝为例,证明了溶解动力学是由两个物理过程之间的竞争动力学,分别与金属-金属键断裂和离子迁移之间的竞争动力学。
计算方法
在整个工作过程中,作者考虑了一个无氧化物的铝(Al)表面,这对于理解低pH条件下或溶液质量传输被抑制的缝隙中的腐蚀特别相关。所有的计算都使用基于网格的投影增强波(GPAW)代码中的DFT实现,并具有真实空间网格基集,采用PBE交换关联函数进行电子计算,真空度设置为18Å。晶格常数优化采用21 × 21 × 21的 k点网格进行,表面计算采用4×4×1网格进行。此外,我们选择研究常见的Al(111)表面,该表面采用3×3×3板建模,其中板的最底层在体位置受到约束。在与表面垂直的方向上,平板的周期性图像之间的距离至少为50 Å。采用溶剂化凝胶法进行了恒定势表面计算,在第一个离子溶剂化壳层中对显式水分子进行溶解能计算。
结果与讨论
图1显示了原子对原子的溶解过程是如何从一个完整的Al(111)表面进行到一个溶剂化的Al3+水配合物。以一个外加原子为例,溶解过程可分为两个步骤:(i)打破金属-金属键,形成全离子溶剂化,用反应I1→I2表示;(ii)离子通过EDL迁移到本体溶液中,用I2→FS表示。溶解动力学由这一过程的演化决定,而溶解的热力学驱动力可以定义为固相中铝原子(IS)向浸在溶剂中的+3离子转变的总反应能。

图1 从Al(111)表面出发的原子逐溶解的反应示意图,以初始吸附原子为例
在继续研究表面及其相关动力学之前,首先计算了Al/Al3+氧化还原电位,这为验证我们的模拟结果提供了方便的参考。电势是通过计算反应能来计算的,反应能与大块金属内部的原子跃迁到浸入溶剂中的Al3+水合离子络合物有关,并伴有三个电子转移到金属(见支持信息)。得到氧化还原电位的值为- 1.48 VSHE,与实验值-1.68 VSHE基本一致。作者指出,纯隐式溶剂化模型不足以准确描述溶剂化离子。例如,如图2A和图S1所示,仅使用单个显式水分子和隐式溶剂模型导致氧化还原电位为+2.09 VSHE,大大高于实验值。当使用[Al(H2O)n]3+簇来描述溶剂化离子时,这种差异在很大程度上得到了纠正,该簇含有6个明确的水分子,形成了一个完整的溶剂化壳。这证实了结合六个明确的水分子形成第一个水化壳以及隐性溶剂化可以合理地用于评估金属的溶解能量。图2A中的结果主要关注Al/Al3+氧化还原电位作为整体性质。实际上,溶解是一种表面现象,它是由溶解原子的化学键局部决定的(图1)。这又取决于多种因素,如表面取向、晶界和缺陷的存在以及特定的溶液环境。在此基础上,作者进一步研究了不同配位数的Al原子的氧化还原电位。为了简单起见,将重点放在无缺陷的单晶Al(111)表面,并考虑表面原子产生最低和最高的配位数。这些物质的氧化还原电位的计算如图2B和C所示。电位是通过计算图1中初始状态(is)和最终状态(FS)之间的能量差得到的。正如预期的那样,溶解表面原子所需的能量(0.41 V)比溶解表面原子所需的能量少得多。I1 |Z
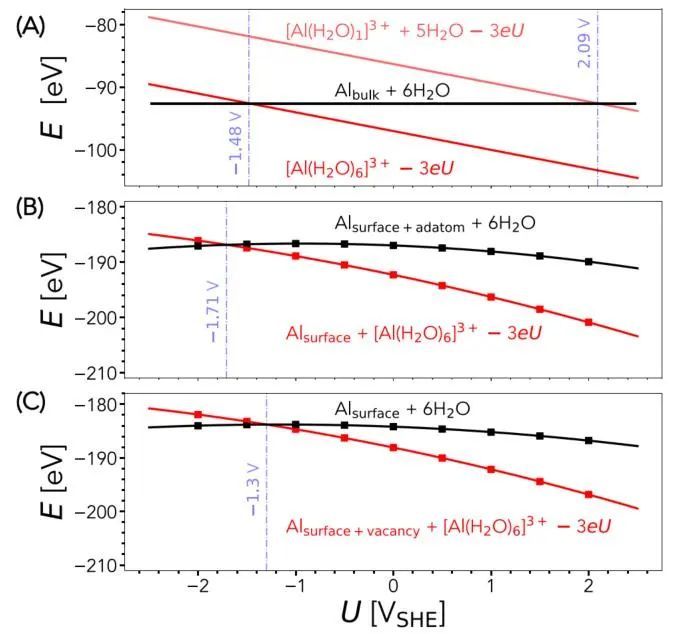
图2 Al表面的溶解反应能
除了Al/Al3+氧化还原电位外,模拟方案还可以扩展到在恒定电位下的溶解动力学。这里作者关注附加原子,如图1所示,溶解过程可以直观地分为两个反应阶段:金属-金属键的断裂和形成一个完整的离子溶剂化(I1→I2);以及离子-水配合物通过EDL(I2→FS)迁移到本体溶液中。在这里,I1态使用5个显式水分子,它们代表部分溶剂化壳,一个额外的水分子在隐式溶剂深处,而I2态的离子溶剂化使用6个显式水分子。I1和I2几何图形代表了[Al(H2O)5 ] 3+和[(H2O)6 ] 3+配合物的I1 |Z和I2 |Z所获得的最小能量结构,分别如图3所示。
在外加电位存在下,通过改变[Al(H2O)5 ] 3+和[(H2O)6 ] 3+配合物的位置,得到了I1 |Z和I2 |Z沿表面法向的势能分布图。在每一步中,优化这些配合物的构型,并将配体固定在Z方向上。通过对图S3所示结构进行优化,得到了I1 |Z=0结构,其中不允许吸附原子在与表面垂直的方向上松弛。与I1→I2步骤相关的过渡态和对应的反应势垒(ΔE‡diss)近似为I1 |Z和I2 |Z构型势能分布的交集。这样,由于第6个水分子的存在,与离子溶剂化的临时重构相关的动力学不包括在模拟中。最后,通过I2 |Z的势能分布图,得到了与I2→FS相关的过渡态及其对应的反应势垒(ΔE‡ion)与表面距离的函数关系。图3显示了- 1 VSHE的典型应用电位下,金属配原子与表面距离的函数图。注意到I2 |Z在长距离处能量的缓慢收敛是由于[(H2O)6] 3+配合物与带电电极之间的静电相互作用(见图S4),即,它依赖于静电势。为了减轻这种缓慢的收敛并保持热力学一致性,分别在恒电位和恒电荷+ 3下处理电极和水离子络合物,得到I2 |Z的最终态(FS)。

图3 在-1 VSHE处的溶解动力学作为吸附原子和Al(111)表面之间的距离的函数
根据这一模拟方案,作者进行了Bader电荷分析,以阐明溶解金属的电荷状态的空间演化。I1 |Z和I2 |Z几何形状与表面距离的函数关系分别如图4A和B所示。发现,由于与外显水的相互作用和初始金属-金属键的拉伸,吸附原子(红线)在I1 |Z=0构型中已经表现出阳离子特征,其电荷状态约为+1.1。正如预期的那样,随着金属-金属键的继续拉伸,水合吸附原子变得越来越氧化,直到键完全断裂。如图4A所示,一旦金属-金属键断裂,电荷大小的下降是由于I1 |Z进入溶液时没有考虑到完整的六水溶剂化壳层。另一方面,在I2 |Z中具有完整第一水化壳层的Al配合原子(图4B),当离子配合物从以金属-金属相互作用为主的状态转变为以金属-溶剂相互作用为主的状态时,产生+2的电荷状态。这表明,随着金属的溶解,它首先演变成+2状态,然后过渡到预期的+3状态;当金属离子迁移到体溶液中时,后者出现外层电子转移现象。
根据电荷密度分析,可以解释离子靠近表面的低电荷状态,当溶剂化离子靠近金属表面时,显示出高度的离域电荷,导致电子从表面溢出到金属离子中。对溶解离子的电荷态演化的研究结果与马库斯理论一致,该理论认为多价离子的溶解涉及一系列单电子步骤,因为同时转移多个电子所需的重组能要高得多。作者的分析还指出,溶解和离子迁移机制之间的电荷转移反应具有根本不同的性质,分别由I1 |Z和I2 |Z构型表示。对于前者,金属-金属键主导电荷转移,导致溶解金属中心和累积水合金属离子配合物的电荷状态相似,分别由图4A中的红色和蓝色线表示。另一方面,由于离子在I2构型中完全溶剂化,金属中心的电荷状态保持稳定在≈2.6,而溶剂化配合物中的其余电荷则在周围的水分子中离域(图4B)。还发现,正是这些水分子和金属表面之间的电荷转移,导致整个离子络合物在从表面迁移时,电荷态增加到+3。
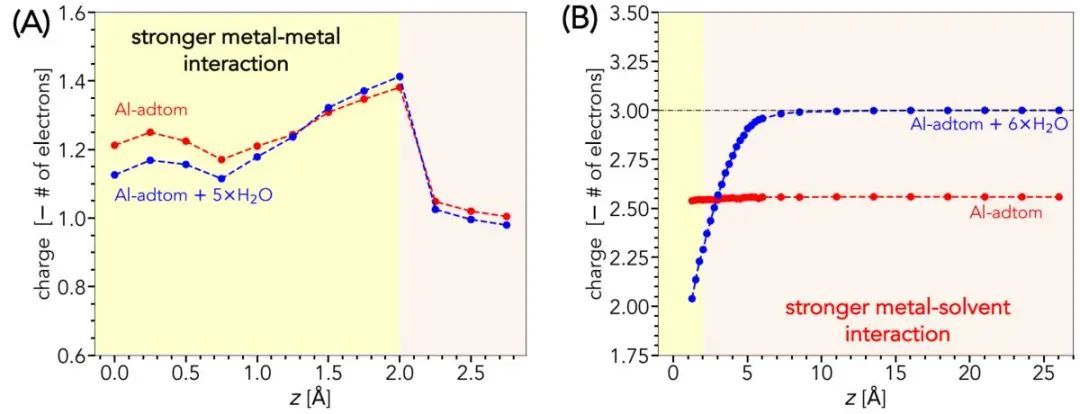
图4 -1 VSHE的I1 |Z和I2 |Z构型作为原子和Al(111)表面之间距离的Bader电荷分析
整个溶解过程的能量学如图5所示,图1中所示的与每一步相关的反应能差与施加的电位(U)呈简单的线性关系。每条曲线的斜率是与该步骤相关的沿反应坐标的电荷转移,由SJM得到。例如,从IS到FS的整个反应产生斜率为~ 3(黑线),这与整个Al溶解过程中总共转移了3个电子相一致。对热力学剖面的考察揭示了重要的附加发现。特别是,从Al原子到部分溶剂化离子(IS→I1,品红色线)的转变总是能量上的有利。另一方面,在非常低的电势下,通过破坏金属-金属键(I1→I2,绿线)形成完整的溶剂化壳层和溶剂化离子通过EDL (I2→FS,蓝线)迁移到体溶液中是热力学上不利的;它们分别在- 1.0 VSHE和- 1.5 VSHE左右变得有利。最后,从纯热力学的角度来看,在采样电位范围内,I1→I2是溶解过程中最困难的步骤。如果考虑动力学的影响,情况就更有趣了。特别是,Al溶解可以由两个动力学限制过程中的任何一个控制,这两个过程与金属-金属键断裂和离子通过EDL迁移有关(图5B,分别为红色和蓝色线)。与热力学相反,动力学势垒对电势表现出更非线性的行为。在低于- 1.7 VSHE的电位下,离子迁移是限速过程,而在更高的电位下,通过打破金属-金属键形成完整的离子溶剂化的过程占主导地位。总的来说,这些结果表明溶解动力学对电极电位的复杂依赖,指出了在涉及电化学溶解的模拟中考虑电位偏置影响的重要性。
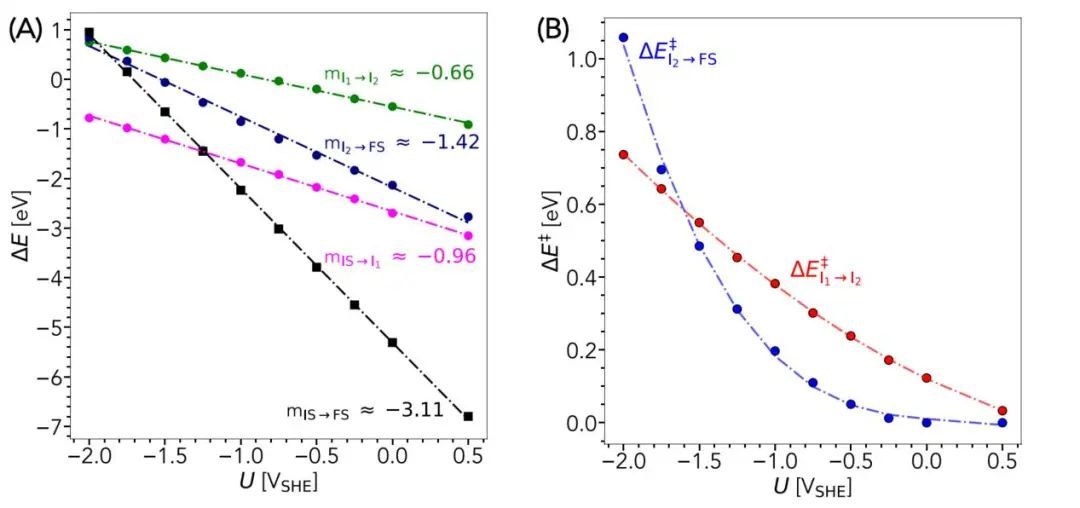
图5 (A)反应能和(B)反应势垒的电势依赖性
结论与展望
总之,作者提出了一个简单的基于DFT的模拟协议,通过巨正则来预测恒定偏置势下的溶解动力学、电荷转移和热力学。应用于金属铝作为一个演示案例,我们的方法被用来证明铝的溶解可以由两个潜在的动力学限制过程中的任何一个来控制。这些过程与金属-金属键断裂和离子通过电双层迁移有关。这些效应包括熵效应,以及与水的输运和离子溶剂化的临时重构相关的动力学,这可能在马库斯理论的背景下高度相关。同样,还需要进一步的努力来了解隐式溶剂化模型的选择以及用来表示离子溶剂化的显式水分子的数量如何影响溶解动力学。
文献信息
Sharma, S., Zagalskaya, A., Weitzner, S. E., Eggart, L., Cho, S., Hsu, T., ... & Wood, B. C. (2023). Metal dissolution from first principles: Potential-dependent kinetics and charge transfer. Electrochimica Acta, 437, 141443.
